





5.GMP製造のバリデーション
~理工学、文系ともベンチャー立ち上げの機会あり~
再生医療が今後大きく発展すると考えられます。その主人公である細胞を医薬品とするにはGMP製造が欠かせません。GMP製造には多額の費用が掛かります。 考え方によっては、これは、ベンチャー創設の良い機会と捉える事ができます。
GMP製造ではどの様な作業があるのか、その詳細は広く知られていないと思います。医薬品開発を行っていた経験から、その内容の内、主だったものについて紹介 します。
GMP製造では、バリデーションが重要です。確かであることを文書で証明する作業と言い換えることが出来ると思います。記録チャート、写真なども、証拠の一部 です。理工系、文系の別なく、多くの人が腕を振るえる分野なのです。
ここでは、1、細胞の大量培養、2、培地の滅菌、3、オートクレーブ滅菌器、4、注射用水製造装置、5、HVAC(Heating, Ventilation, and Air Conditioning) およびクリーンベンチについて、どの様なバリデーションが行なわれているか、具体的に紹介します。ベンチャー創設のヒントを掴んで下さい。ITベンチャーもきっと 立ち上げられる筈です。
1 細胞の大量培養
細胞を大量に得る方法としては、液体培地中で浮遊培養できる細胞なら、タンク培養を行う事が現実的です。個体に付着しなければ増殖しない様な細胞では、 ビーズ、メッシュなどを活用した装置を組む必要があります。シャーレを何千枚・何万枚も並ベて培養する事は現実的ではないのです。
いずれにしても、同じ性質の細胞が再現性良く造れる事を証明し、文書に残さなくてはなりません。研究・開発段階から記録を残す習慣が大切です。実験ノートは 改竄が出来ないような工夫が必要です。電子記録も同様です。
タンク培養の条件として、若し、撹拌が必要なら、その回転数・時間を、また、温度管理が重要ならその温度と時間を、記録に残すのです。
タンク培養では温度が重要ですが、その時の温度に勾配があればその変化も記録しなければなりません。計測・記録に用いる温度計は、校正されている事が信頼性の 必要条件の1つです。なお、校正の頻度は、重要工程では半年に1回、そうでないものでも、1年に1回は行います。また、世界標準に繋がる証明書を入手しなくては なりません。温度センサーにK型熱電対を使えば、その起電力の検定を、白金センサーなら抵抗の検定も必要です。CO2など気体や培地など液体を加える場合は流量の記録 が必要です。勿論、それら計器の校正も必要です。
この沢山の校正が効率的に出来るよう改善するだけでもベンチャーが創出出来る可能性があります。
作業の基本は製造基準書に記載します。製造に当たっては、毎回、それに準拠した製造指図書を作成します。その指図書は左ページに記載され、製造記録は右側に なります。指図に従い、誰が何時、どのように製造したかを記録しながら製造していくのです。重要な箇所では、作業者のサインも必要です。この沢山の記録書類の作成 は、理工系・文系の区別なく、担当出来ますし、実際、GMP製造はチームの総力を挙げての作業になります。
因みに、原料についても保証が必要です。細胞培養ですから、水、アミノ酸、糖類、ミネラル、場合によっては血清などが原料となりますが、それら総ての原料の 品質を確かめなくてはなりません。純度、夾雑物の有無とか、混入が避けられないなら、その割合とか、不安定な物にあっては保存期間とか色々あります。確認したら 記録に残します。文書作成作業、文書管理は大変です。
それらの記録が揃っているか、基準書は正しいか、後に述べますが、各種バリデーションが頻度を正しく守って行われているか、出荷に際しては規格試験に合格して いるか等、信頼性を保証する部門の活躍も必要です。この部門の権限は独立していなければなりません。経営者と対等の権限となっているのです。この様な品質保証の 仕事、新人製造担当者の教育訓練の仕事もあります。FDA査察官は教育訓練記録も調べます。
2 培地の滅菌
細胞培養用の培地は熱に弱いので、オートクレーブなどの加熱滅菌器で滅菌できません。「ろ過滅菌」と言う手法で細菌をろ過します。因みに、ウイルスの昔の 名称は、ろ過性病原体です。この名称から分かるように、細菌サイズ用のフィルターではろ過出来ません。培地成分として良く使われる血清などは、抗体検査など適切な 方法で、支障となるウイルスが入っていない事を確認して使います。
「ろ過滅菌」は単に除菌出来る膜を通せば良いのでは無く、ろ過した後、使用した膜が破れておらず、正しくろ過が出来た事を証明します。完全性試験器という 名称の機器が有り、これで、膜の破損が無かった事を証明します。膜が破れていた時の耐圧と完全であった時の耐圧には差が有り、この計器で判別できるのです。勿論、 行った作業毎に証拠を文書または図(チャート)で残さなくてはいけません。日付、実施者のサインも必要です。

3 オートクレーブ滅菌
細胞は、出来上がってからでは滅菌が出来ません。無菌操作で調製する必要が有ります。従って、用いる器具、容器、計測器のセンサー部分など、細胞と接触 するものは予め総て滅菌しておかなければなりません。それにはオートクレーブが良く使われます。または、装置を組み立ててから一式で滅菌するSIPと言う方法も あります。
原則は一緒ですので、簡単なオートクレーブ滅菌の例について述べます。
1、設計の適格性を検証します(DQ)。
2、据え付けの適格性を検証します(IQ)。
3、運転が正しく行えるかどうか検証します(OQ)。
この段階では被滅菌物を負荷しません。
3-1、検査箇所を決めます。缶体の大きさにより増減しますが、試験室タイプの大きさで9か所調べます。
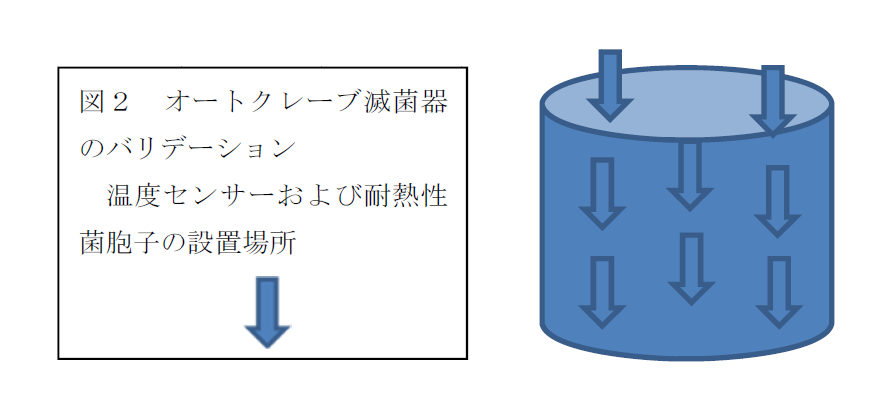
3-2、指定した9箇所総てに温度センサーを設置し、記録計に繋ぎます。
3-3、指定した箇所総てに標準耐熱性菌の胞子100万個をセットします。
3-4、既定の温度・時間運転し、温度、圧力を記録します。チャート取得。
3-5、胞子を取り出して培養します。
チャートが所定の温度。圧力に一定時間達している事、検査した標準耐熱性菌が総て死滅している事を確認します。
オートクレーブは9本以上の白金センサーが通る機密式の穴を備えている必要があります。研究室用のオートクレーブはその穴が無いのが普通です。 改造が必要です。
4、滅菌したいものが確実に滅菌できることを検証します(PQ)。
4-1、滅菌したいものを使用時の形状にします。滅菌缶にセット、または滅菌バッグなどにシールするのです。
4-2、最大負荷パターンを予め予備実験で調べ、写真に撮っておきます。
缶内にセットするパターンは、この最大負荷パターンを超えないようにセットします。次いで3のOQと同じように運転します。
センサーと標準菌の検査箇所は、「数」は同じとしますが、「箇所」は、空気が抜けにくいなど、最も滅菌され難い箇所が含まれるよう設定します。
査察官に科学的・論理的に説明出来るようにしておくためです。
4-3、負荷した状態で運転します。チャートは重要です。
4-4、温度・圧力が滅菌に必要な状態に達している事を確認します。
4-5、胞子を取り出して培養します。菌の増殖が認められてはなりません。
3のOQおよび4のPQにおいて、無菌性を調べる培地は、標準菌が生育する性能を有していなければなりません。具体的には、その時使用する培地の√n+1を無作為 に選び、標準菌培養液を接種します。接種した菌量の50%以上が生育する事を確認します。
4 注射用水製造装置
医薬品製造において注射用水(WFI:Water for Injection)は重要です。細胞医薬品の場合、水は原料の一部ですし、使う器具・装置の洗浄に必要です。細胞培養の 場合、グラム陰性桿菌の膜成分として知られるLPSは混入してはなりませんが、その汚染対策としても、注射用水によるリンスは有用です。
このWFIは国によって基準が異なります。製法の違いとしては、日本では、蒸留法、逆浸透膜法、限外ろ過膜法のいずれの方法で造っても良いのですが、米国や ヨーロッパ(EU)では蒸留法だけ、または蒸留法と逆浸透膜法だけが認められています。水質基準も異なります。日本、FDAおよびEU当局の基準をすべて満たすWFIの製造 は、検査が多くなり、コストが掛かる事になります。
WFI製造装置は、日本では、栗田工業、日本濾水機工業など多くあります。外国では蒸留法が主体です。つまり、多重効用缶を有する装置が主流です。この製造装置 は数1000万円あるいは億単位の装置です。
このWFIを製造する装置の材質と構造としては、下記の事が知られています。
○材質:SUS316L
○構造:二重管版構造
○管内腔、タンク表面処理:鏡面処理、電解研磨
○継手:サニタリー継手(ネジ禁止)
○バルブ:ダイヤフラムバルブ(ボール・タップ バルブ禁止)
○配管勾配:1/100以上
○ドレンの位置:最下部となる総ての位置
○死腔:管直径(d)の6倍以下(6d以下でないと殺菌できないからです。従って、WFI装置は循環構造をとります。)
○最も温度が低くなる点に監視用センサー設置
この装置により製造されるWFIが合格する基準は、若し、米国FDAの場合であれば、米国薬局方(USP)に適合している事を証明するのです。 具体的には次のような検査をします。
・総てのユースポイント(蛇口)から5週間にわたってサンプリングする。
・採取頻度は0時間、1h、半日、翌日、2日後、3日後、1週間後、以後毎週など等比級数的に設定
・総てWFI基準に合格する事を確認
記録は総て残さなくてはいけません。無菌試験法、リムルス・テストなども水質検査に含まれますが、これらの試験法も、FDA査察を対象とするなら、USP記載の方法 で行います。試験が的確である事を証明するためです。なお、リムルス・テストはLPSのテストのことで、陽性対照に米国標準品(リファレンス・スタンダード・ エンドトキシン)を購入し、それを基準として、日々の検査に用いる自分達用のスタンダード・エンドトキシンの力価を求めておきます。
とにかく、世界基準と繋がっている事が重要です。この様に、水関係は費用の掛かる分野です。ここでもベンチャーの可能性が大きいのです。 水商売ですから儲かるかも(冗談でなく)。
5 HVACおよびクリーンベンチ
製造工場の空気環境は重要です。特に、細胞を医薬品にするなら、無菌は必須条件です。クリーンルーム、クリーンベンチが必要になってきますが、その設計や 運転、バリデーションには厳しいものがあります。
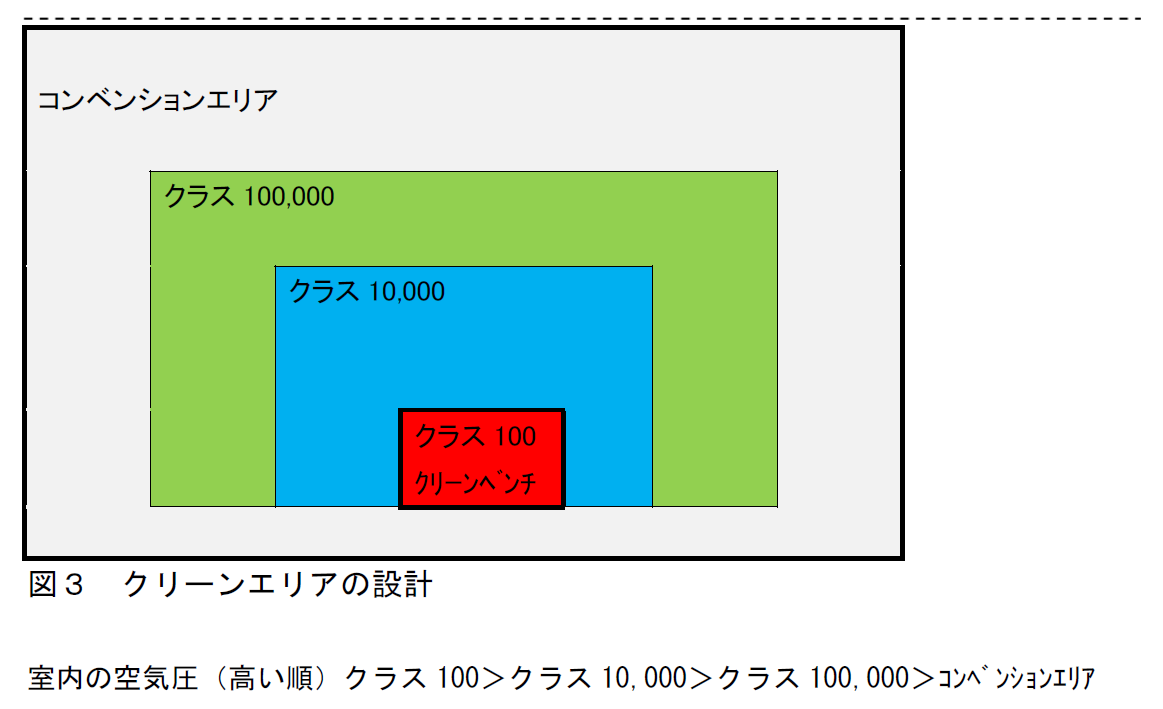
図3はクリーンエリア設計の概念を示したもので、清浄な度合いが高まるほど陽圧にし、汚染が起きないようにします。危険な病原体を扱う施設とは真逆になり ます。
また、清浄度の高いクラスをいきなり設けず、段階を付けて設置します。
クリーンルームの素材は洗剤、消毒液、殺菌剤などに耐えられる素材とし、表面は平滑で、製造の前後の無菌化が確実に行えるようにします。
また、製造中は微生物学的なモニタリングをして製品がクリーンに製造出来た証拠とします。モニタリング項目としては、空中浮遊菌数、空中微粒子数、付着菌数、 落下菌数、無塵衣を着た作業者の付着菌数、特に、装着した滅菌手袋の付着菌数などです。
菌は検出されないはずですが、作業者のガウニング(無塵衣の着方)や滅菌手袋装着方法に不備があれば、また、クリーンルームの故障などが有れば、時として、 菌が検出されます。その場合は、検出された菌を、例えば、APIシステムなど、市販のキットを用いて同定します。その結果を基に、製品の汚染の可能性、作業者の ガウニング・滅菌手袋装着の教育訓練は適切であったか、クリーンルームの機能は損なわれていなかったかなど検討します。製品汚染の可能性があれば出荷停止 です。
この他、作業環境が汗だくになるような環境ではまともな製品が出来ませんので、温湿度も調えます。温湿度、室間差圧等は連続的にモニターし、チャートに記録 します。勿論、その計器の校正も定期的に行います。
また、各クリーンルームは清浄度に応じたHEPA(「High Efficiency Particulate Air Filter」)フィルターを備えており、それらの性能を定期的に調べます。 最も厳しい検査はクラス100のクリーンベンチです。製造中のモニタリングに加え、6か月に一度の割合で、垂直層流であること、HEPAフィルターに試験的に1立方フィート 当たり微粒子100万個以上負荷し、その99.97%以上が捕捉出来る事、ピンホールなど無い事、空気中に細菌が無い事を調べます。
空気サンプリング装置がザルトリウス社、ミリポア社などから販売されています。 これらバリデーションの新装置を開発するベンチャーも考えられますが、これらバリデーションを請け負う業務でベンチャー企業を興す事も十分考えられます。
先ずは現場を体験し、改善点を見つけ出す事です。
以上、バリデーションの実際について具体的に述べました。GMP製造は大変であることをご理解戴ければ本望です。と同時に、GMP製造の中にベンチャー創設の 可能性が秘められている事も理解して戴ければ幸いです。
文責&連絡先:国立大学法人 金沢大学 先端科学・イノベーション推進機構
客員教授 長江 英夫 博士(工学)、獣医師
nagaehi@staff.kanazawa-u.ac.jp 携帯:090-7670-5508